Welcome to the NIPT course
This course is designed to share knowledge about NIPT only and is based on the book Noninvasive Prenatal Testing (NIPT).
background
1997 年,Lo 等人证实,在怀孕期间,孕妇的血浆和血清中可以看到胎儿 DNA [1]。随后的研究证明了胎儿 DNA 在妊娠期的变化[2]以及在分娩后的快速清除[3]。研究发现,循环中的胎儿 DNA 由短 DNA 片段组成,其大小分布短于母体血浆中循环中的母源 DNA [4]。
在诊断学方面,我们见证了这一年轻领域在过去二十年中的飞速发展。本书总结了这一时期的重要发展。早期的工作主要集中在检测胎儿从父亲那里继承的、而怀孕母亲基因组中不存在的 DNA 序列,如男性胎儿的 Y 染色体[5]、RhD 阴性母亲携带的 RhD 阳性胎儿的 RHD 基因[6,7],以及胎儿从父亲那里继承的、但在母亲基因组中不存在的突变[8]。
最近,微流控数字 PCR、液滴数字 PCR 和大规模并行测序等新技术的应用也加入了这些早期应用的行列。因此,单基因疾病的无创产前检测现已扩展到阐明常染色体隐性遗传疾病[9-11]和性连锁疾病[12,13]中胎儿的父系和母系遗传。
过去几年最受关注的无创产前检测领域是利用大规模平行测序技术检测胎儿染色体非整倍体[14,15]。自 2011 年首次大规模临床试验证明该技术的稳健性以来[16,17],该技术已在全球数十个国家迅速引入临床实践。在检测染色体非整倍体之后,这种方法也被迅速用于检测亚染色体畸变[18,19]。
母体血浆中胎儿 DNA 的诊断潜力的最终体现,或许是证明了可以从母体血浆中确定整个胎儿基因组[20-22]。在这些早期研究之后,最近又有人利用较新的测序和生物信息学方法阐明了所谓的第二代无创胎儿基因组[23]。通过这些方法,可以从母体血浆中对胎儿的新基因突变进行全基因组检测,还可以确定胎儿的母体遗传,其分辨率比之前的尝试高出约两个数量级[23]。
Hence, one can see from the above that developments of the diagnostic applications of fetal DNA in maternal plasma have been most remarkable over the last two decades. However, there is still much to be learnt. For example, the biological characteristics of circulating fetal DNA still remain to be completely elucidated. Emerging areas of investigation include the relationship between circulating DNA and nucleosomal structure [24,25], the existence of preferred plasma DNA fragment endpoints[23], and the tissues of origin of circulating DNA[26,27]. Finally, a thought-provoking and unresolved problem is whether circulating fetal DNA has any biological or pathogenic functions. Hence, the next two decades will certainly be very exciting. 由此可见,过去二十年来,母体血浆中胎儿 DNA 的诊断应用取得了长足的发展。然而,我们仍有许多东西需要学习。例如,循环胎儿 DNA 的生物学特征仍有待完全阐明。新的研究领域包括循环 DNA 与核糖体结构之间的关系[24,25]、血浆 DNA 片段终点的存在偏好性[23]以及循环 DNA 的组织来源[26,27]。最后,循环胎儿 DNA 是否具有生物或致病功能也是一个发人深省且尚未解决的问题。因此,未来二十年肯定会非常精彩。
References
[1] Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, et al. Presence of fetal DNA in ma- ternal plasma and serum. Lancet 1997;350(9076):485–7.
[2] Lo YM, Tein MS, Lau TK, Haines CJ, Leung TN, Poon PM, et al. Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet1998;62(4):768-75.
[3] Lo YM, Zhang J, Leung TN, Lau TK, Chang AM, Hjelm NM. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet 1999;64(1):218–24.
[4] Chan KC, Zhang J, Hui AB, Wong N, Lau TK, Leung TN, et al. Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem 2004;50(1):88–92.
[5] Costa JM, Benachi A, Gautier E. New strategy for prenatal diagnosis of X-linked disorders. N Engl J Med2002;346(19):1502.
[6] Faas BH, Beuling EA, Christiaens GC, von dem Borne AE, van der Schoot CE. Detection of fetal RHD- specific sequences in maternal plasma. Lancet 1998;352(9135):1196.
[7] Lo YM, Hjelm NM, Fidler C, Sargent IL, Murphy MF, Chamberlain PF, et al. Prenatal diagnosis of fetal RhD status by molecular analysis of maternal plasma. N Engl J Med 1998;339(24):1734–8.
[8] Chiu RW, Lau TK, Leung TN, Chow KC, Chui DH, Lo YM. Prenatal exclusion of beta thalassaemia major by examination of maternal plasma. Lancet 2002;360(9338):998–1000.
[9] New MI, Tong YK, Yuen T, Jiang P, Pina C, Chan KC, et al. Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. J Clin Endocrinol Metab 2014;99(6):E1022-30.
[10] Hui WW, Jiang P, Tong YK, Lee WS, Cheng YK, New MI, et al. Universal haplotype-based noninvasive prenatal testing for single gene diseases. Clin Chem 2017;63(2):513–24.
[11] Barrett AN, McDonnell TC, Chan KC, Chitty LS. Digital PCR analysis of maternal plasma for noninvasive detection of sickle cell anemia. Clin Chem 2012;58(6):1026–32.
[12] Tsui NB, Kadir RA, Chan KC, Chi C, Mellars G, Tuddenham EG, et al. Noninvasive prenatal diagnosis of hemophilia by microfluidics digital PCR analysis of maternal plasma DNA. Blood 2011;117(13):3684-91.
[13] Hudecova I, Jiang P, Davies J, Lo YMD, Kadir RA, Chiu RWK. Noninvasive detection of F8 int22h-related inversions and sequence variants in maternal plasma of hemophilia carriers. Blood 2017;130(3):340-7.
[14] Chiu RW, Chan KC, Gao Y, Lau VY, Zheng W, Leung TY, et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci USA 2008;105(51):20458-63.
[15] Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci USA 2008;105(42):16266–71.
[16] Chiu RW, Akolekar R, Zheng YW, Leung TY, Sun H, Chan KC, et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: large scale validity study. BMJ 2011;342:c7401.
[17] Palomaki GE, Kloza EM, Lambert-Messerlian GM, Haddow JE, Neveux LM, Ehrich M, et al. DNA sequenc- ing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med2011;13(11):913-20.
[18] Srinivasan A, Bianchi DW, Huang H, Sehner t AJ, Rava RP. Noninvasive detection of fetal subchromosome abnormalities via deep sequencing of maternal plasma. Am J Hum Genet 2013;92(2):167–76.
[19] Yu SC, Jiang P, Choy KW, Chan KC, Won HS, Leung WC, et al. Noninvasive prenatal molecular karyotyp- ing from maternal plasma. PLoS One 2013;8(4):e60968.
[20] Lo YMD, Chan KCA, Sun H, Chen EZ, Jiang P, Lun FMF, et al. Maternal plasma DNA sequencing reveals the genome-wide Genetic and mutational profile of the fetus. Sci Transl Med 2010;2(61):61ra91.
[21] Kitzman JO, Snyder MW, Ventura M, Lewis AP, Qiu R, Simmons LE, et al. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med 2012;4(137):137ra176.
[22] Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR. Non-invasive prenatal measurement of the fetal genome. Nature 2012;487(7407):320-4.
[23] Chan KC, Jiang P, Sun K, Cheng YK, Tong YK, Cheng SH, et al. Second generation noninvasive fetal ge- no me analysis reveals de novo mutations, single-base parental inheritance, and preferred DNA ends. Proc Natl Acad Sci USA 2016;113(50):E8159–68.
[24] Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016;164(1-2):57-68.
[25] Straver R, Oudejans CB, Sistermans EA, Reinders MJ. Calculating the fetal fraction for noninvasive prenatal testing based on genome-wide nucleosome profiles. Prenat Diagn 2016;36(7):614–21.
[26] Sun K, Jiang P, Chan KC, Wong J, Cheng YK, Liang RH, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci USA 2015;112(40):E5503-12.
[27] Guo S, Diep D, Plongthongkum N, Fung HL, Zhang K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat Genet 2017;49(4):635-42.
Noninvasive Prenatal Testing (NIPT). https://doi.org/10.1016/B978-0-12-814189-2.00001-3 3 ©2018 Elsevier Inc. All rights reserved.
INTRODUCTION: EVOLUTION OF SEQUENCING TECHNOLOGY
The term “next-generation sequencing” (NGS) begs the questions of what “first-generation sequencing” was and how NGS is both similar to and different from its predecessor. Sanger developed the first generation of DNA sequencing in the 1970s[1,2]. His eponymous sequencing approach is an in vitro adaptation of the cellular replication machinery that cleverly leverages unextendable DNA bases. These modified bases are introduced at low concentration in a reaction minimally containing (1)a high concentration of extendable bases, (2)the single-stranded DNA template to be sequenced, (3)a short oligonucleotide primer complementary to the template onto which new bases could be synthesized, and(4) DNA polymerase enzymes that execute the extension reaction. Early Sanger sequencing experiments involved four independent reactions, each containing a single type of unextendable base (A, T,G, or C). Whenever a polymerase randomly incorporates one of the unextendable bases into the nascent DNA molecule (e. g., an unextendable G in the nascent strand incorporated opposite a C in the template), further synthesis would terminate, yielding a truncated copy of the template. Critically, since all nascent strands anchor from the same oligonucleotide primer, the position of extension termination— and hence the length of the nascent DNA strand— is a direct proxy for the base at the 3' end of the molecule. By using gel electrophoresis to resolve the respective lengths of terminated molecules in each of the four reactions, it is possible to infer the sequence of the entire template.
Sanger sequencing became slightly more scalable with the introduction of unextendable bases that were uniquely dyed( Fig. 1). Rather than achieving base-specific information by partitioning into four reactions, a capillary electrophoresis instrument coupled with a fluorescent dye detector could resolve both the relative sizes of fragments and the identity of their terminating bases [3-5]. To criticize these machines as unscalable neglects one of their unmitigated triumphs: they were the workhorses that sequenced the very first human genome in the 1990s [6-9]. However, with a cost in the billions of dollars and with a timeline on the order of years, genome sequencing would remain prohibitive in a clinical setting without a major technological leap.
NGS revolutionized genome sequencing by overcoming many of the limitations of the Sanger technique [10], yet the most pervasive NGS methodology shares much in common with its predecessor. As described in further detail later,NGS also leverages extension termination and fluorescent bases, and it relies upon DNA polymerases that append a single base at a time to a nascent DNA molecule. Indeed, in many respects, an NGS experiment is comparable to performing millions or billions of Sanger reactions in parallel ( hence the NGS moniker“massively parallel sequencing”). This explosive increase in throughput shattered some of the barriers(e. g., cost and turnaround time) that had largely prevented the use of genomics in routine clinical care, and it paved the way for cfDNA-based prenatal testing.
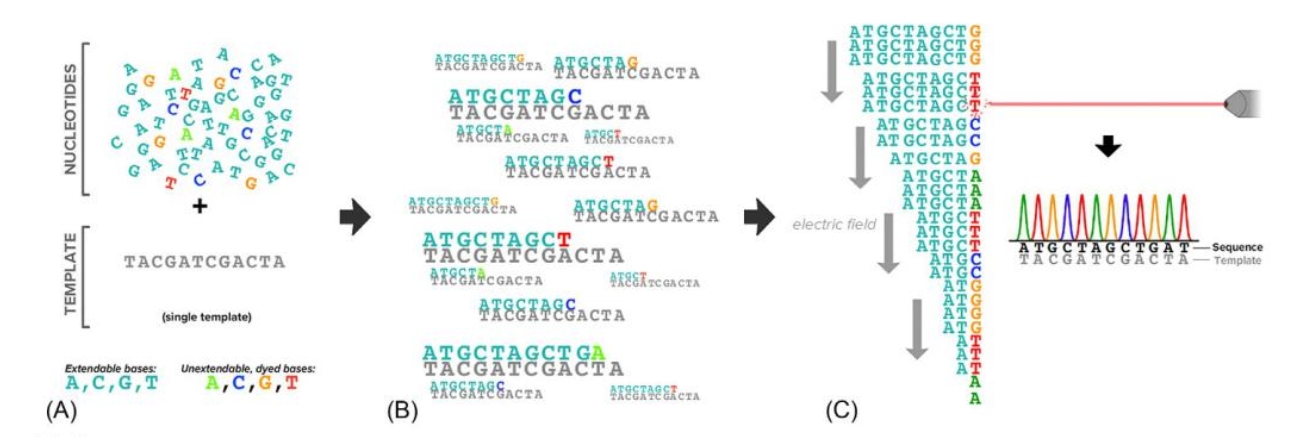
[FIG1]: Overview of Sanger sequencing.(A) The Sanger synthesis reaction contains a nucleotide mix with both extendable bases and a low concentration of unextendable, dyed bases(a DNA primer, DNA polymerase, and buffer are also required an not shown).(B) Extended molecules terminate at different locations with the molecule having the color of the last incorporated base.(C) Arrangement of the terminated molecules in an electric field—DNA is negatively charged— and passage through a capilary equipped with a dye detector identifies fluorescence peaks that reveal the original template's sequence.
How NGS Works
The role of an NGS device is to distill a specially prepared library of DNA molecules into a long text file of sequences, one line for each sequenced molecule. NGS sequencers perform this molecule-to-text mapping across many research and clinical contexts, spanning everything from RNA sequencing in broccoli[11] to ribosome profiling in bacteria [12] to DNA sequencing for NIPT in pregnant women[13]. These applications of NGS are distinguished primarily by the steps upstream of DNA being injected into the sequencer, termed “library preparation.” Mirroring the diversity of upstream sample preparation methods is a comparably vast range of downstream analyses, one of which is the analysis method for NIPT, covered in detail in Chapter 3. In addition to describing how an NGS machine sequences DNA, this section discusses NIPT-specific workflows upstream and downstream of sequencing.